Міопатія - діагноз неабиякий. У даній статті практикуючий лікар розповідає про види цього багатоликого недуги.
зміст
 «Ми - три брата (31 рік, 29 і 27 років) - з юності страждаємо від одного захворювання - прогресуючої м'язової дистрофії. Всі троє - інваліди. Може, відгукнуться лікарі-фахівці на нашу біду і допоможуть.»
«Ми - три брата (31 рік, 29 і 27 років) - з юності страждаємо від одного захворювання - прогресуючої м'язової дистрофії. Всі троє - інваліди. Може, відгукнуться лікарі-фахівці на нашу біду і допоможуть.»
«У дитини (10 років) прогресуюча м'язова дистрофія Дюшена. лікарі безсилі. Буду вдячна за будь-які рецепти, поради.»
«В онука (3 роки) раптово стали відмовляти ноги і з часом стає все гірше. Лікарі ставлять різні діагнози і нічого не можуть зробити. Допоможіть доброю порадою.»
Це витяги з листів людей, що зіткнулися з таким серйозним захворюванням як міопатія. Що ж таке міопатія? Давайте спробуємо класифікувати цей багатоликий недуга.
Міопатія і її види
Міопатії представляють групу нервово-м'язових захворювань, які проявляються стомлюваністю, слабкістю м'язів, зниженням м'язового тонусу, атрофією м'язів. Міопатії, в залежності від причинного фактора, поділяються на прогресуючу спадкову м'язову дистрофію, ендокринні міопатії (захворювання залоз внутрішньої секреції) і метаболічні міопатії (порушення обміну речовин).
Поговоримо про прогресуючу спадкової м'язової дистрофії. Цей вид міопатії характеризується атрофією м'язів за рахунок руйнування м'язових клітин в зв'язку з нестачею спеціального білка, який зміцнює структуру м'язових волокон. Цей білок виробляється під контролем спеціального клітинного гена, що розташований на 6-й хромосомі людини, і при дефекті цього гена настає поступове руйнування оболонок м'язових клітин з наступним омертвением м'язових волокон.
Цей дефектний ген передається у спадок, якщо в роду був шлюб між родичами. Зміни гена в 30% випадків відбувається в результаті мутації, тобто в цих випадках шлюб між родичами - ні при чому. Хвороба передається у спадок з імовірністю 50%, якщо один з батьків дитини хворий. Він пов'язаний з жіночою статевою хромосомою і передається, як правило, синам, хоча самі жінки можуть і не хворіти. Атрофії піддаються м'язи плечового пояса рук, спини, тазового пояса і ніг.
Залежно від локалізації хвороби, віку, тяжкості захворювання виділяють різні форми м'язової дистрофії. Так, юнацька форма Ерба-Рота виникає у віці 10-20-ти років, коли непомітно з'являється атрофія м'язів плечового пояса і рук, а потім - тазового поясу і ніг. Під час ходьби хворий перевалюється з випнутих вперед животом і відсунути назад грудною кліткою. Щоб встати з положення лежачи, хворий повертається на бік і, спираючись руками на стегна, поступово піднімає свій тулуб. Хвороба повільно прогресує.
Дитяча форма м'язової дистрофії Дюшена починається у віці 3-5-ти років з атрофії м'язів тазу, стегон з одночасним потовщенням литкових м'язів гомілки (помилкове потовщення). Поступово атрофуються м'язи плечового пояса і рук. У дітей спочатку порушується хода, а потім виникає складність в пересуванні. У багатьох порушується серцевий ритм за рахунок збільшення розмірів серця. Прогресування захворювання або його злоякісний перебіг у зв'язку з раннім знерухомленням кінцівок призводить до сумного результату. Хворіють, в основному, хлопчики (1 на 3000 народжених). Якщо бути більш точним, хворіють і чоловіки і жінки в рівній мірі. Тільки хвороба Дюшена проявляється у хлопчиків. Дівчата є носіями цього гена.
Але буває і доброякісний перебіг м'язової дистрофії (миодистрофия Беккера), коли захворювання проявляється повільно, особливо у низькорослих дітей. Багато років вони зберігають задовільний фізичний стан і тільки приєднання різних гострих захворювань і травм призводить їх до знерухомлених, виснаження з поганим результатом.
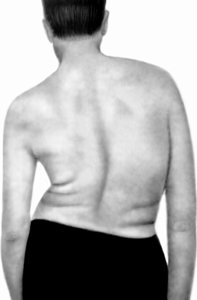
Виділяється плече-лопаточно-лицьова форма міодистрофії, звана Ландузі-Дежерина, яка може бути у віці від 6-ти до 52-х років (частіше в 10-15 років) і характеризується ураженням м'язів обличчя з поступовою наступною атрофією м'язів плечового пояса, тулуба і кінцівок. Ранніми ознаками хвороби є погано змикаються і не закриваються повіки, повністю не змикаються губи, що створює нечітку мова і неможливість надути щоки. Захворювання протікає повільно. Довгий час хворий може пересуватися і зберігати працездатність, а потім через 15-25 років поступово атрофуються м'язи тазового пояса ніг, що ускладнює пересування.
Виділяється також група вторинних прогресуючих м'язових дистрофій, які виникають у зв'язку з поразкою нервів: невральна, спінальна міодистрофії, звані ще аміотрофією.
До невральної відноситься амиотрофия Шарко-Марі, яка характеризується поступовою атрофією дрібних м'язів стоп, потім атрофуються м'язи гомілок і нижньої частини стегон, а м'язи середньої та верхньої частин стегон не змінюються і стегно представляє форму пляшки з шийкою, перекинутого вниз. Потім поступово атрофуються м'язи кистей рук і передпліч. Чи не уражаються м'язи тулуба, плечового пояса і особи. Захворювання виникає у віці 18-25 років, повільно прогресує і стабілізується.
Вроджена спінальна м'язова атрофія Кугельберга-Веландера характеризується поступовою атрофією м'язів рук, ніг, затримкою психічного та фізичного розвитку, деформацією хребта. Хвороба проявляється у віці 8-10-ти років і повільно прогресує.
Прогресуюча амиотрофия Арана-Дюшена починається у віці 25-50-ти років і проявляється атрофією м'язів кистей. Потім поступово атрофуються інші м'язи рук, потім ніг тулуба, в т.ч. міжреберних м'язів, що викликає дихальні порушення, від яких настає смерть.
Вроджена аміотонія (зниження тонусу м'язів) Оппенгейма характеризується слабкістю м'язів у зв'язку з їх недорозвиненням, і м'язова дистрофія їх є вторинною. У новонароджених вона не прогресує, але потрапляння респіраторних інфекцій може викликати запалення, і смерть настає на першому році життя. З віком рухова функція м'язів покращується.
Лікування м'язової дистрофії направлено на уповільнення дистрофічних (руйнують) процесів в м'язах і навіть їх припинення. Однак радикального лікування поки не знайдено. Хоча надія є на генну терапію, яка починає повільно впроваджуватися в медичну практику.